
| La saison 2021-2022 de l’influenza aviaire hautement pathogène (IAHP) (période du 01/08/2021 jusqu’à la date de ce rapport) est marquée par une forte prédominance du sous-type H5N1, qui représente 97,8 % des foyers de volailles et 95 % des cas « autres que les volailles, dont les oiseaux oiseaux sauvages ». Le sous-type H5N8, majoritaire lors de la saison 2020-2021, est identifié sporadiquement. En Europe, la totalité des séquences H5 appartiennent au clade 2.3.4.4b. En France, la saison est marquée par un nombre élevé de génotypes appartenant à un seul sous-type de virus IAHP A(H5N1), dont deux génotypes associés à chacune des deux vagues épizootiques majeures en élevages de volailles : FR1 majoritaire dans le Sud-Ouest (de mi-décembre 2021 à début février 2022), puis FR2 majoritaire dans l’Ouest (de mi-février à mi-mai 2022). |
Pour le comité de rédaction de la Plateforme ESA : Jean-Philippe Amat, Eric Cardinale, Sophie Carles, Julien Cauchard, Céline Dupuy, Sylvain Falala, Guillaume Gerbier, Viviane Hénaux, Renaud Lancelot, Célia Locquet, Carlène Trévennec
Pour le LNR : Eric Niqueux, Béatrice Grasland, Audrey Schmitz, François-Xavier Briand, Sophie Le Bouquin-Leneveu, Axelle Scoizec
Auteur correspondant : plateforme-esa@anses.fr
Le document Sources de données précise la terminologie utilisée aux niveaux européen et international pour déclarer les cas d’influenza aviaire hautement pathogène (IAHP) et la notion de pathogénicité des souches d’influenza au sens de l’OMSA.
Europe
Des analyses phylogénétiques ont été réalisées par le LRUE sur des séquences de virus IAHP de sous-type H5 collectées entre octobre 2021 et mars 2022 et, provenant de dix-neuf États-membres de l’Union européenne, de Moldavie, de Norvège, du Royaume-Uni et de Russie. Ces virus appartenaient quasi-exclusivement au sous-type H5N1 devenu dominant. Les analyses ont montré les éléments suivants :
- la totalité des séquences H5 appartiennent au clade 2.3.4.4b.
- l’analyse des huit segments génomiques indique la présence persistante en Europe du Nord de virus IAHP A(H5N1) et A(H5N8) étroitement apparentés aux virus ayant précédemment circulé en Europe depuis octobre 2020, ces derniers restant toutefois minoritaires.
- la détection de la grande majorité des virus séquencés (environ 96 %) serait la conséquence de nouvelles introductions associées à la migration descendante d’oiseaux sauvages infectés. Au moins vingt nouveaux génotypes ont été identifiés par le LRUE[1], qui résulteraient de multiples événements de réassortiment des virus IAHP entre eux et avec des virus influenza aviaires faiblement pathogènes (IAFP). Dix-neuf génotypes distincts de virus IAHP A(H5N1) (alors qu’un seul génotype avait été caractérisé au cours de la saison précédente depuis octobre 2020) et un génotype de virus IAHP A(H5N2) ont ainsi été caractérisés.
- seuls certains de ces génotypes ont été identifiés dès septembre-octobre 2021 en Russie orientale et centrale, mais la plupart correspondent à des associations de segments non décrites auparavant en Europe ou dans d’autres pays, ce qui suggère la survenue d’événements autochtones de réassortiment (source : EFSA rapport trimestriel décembre 2021-mars 2022, le 31/03/2022).
France
Les premières détections de la saison a été signalées au mois de septembre et confirmées H5N8 dans des basse-cours en lien avec des marchés d’oiseaux vivants en Belgique (BHVSI du 04/09/2021). Le sous-type H5N1, a été détecté à partir du 09/11/2021 dans l’avifaune sauvage dans les départements de la Meuse et de Meurthe-et-Moselle (BHVSI du 16/11/2021), puis le 21/11/2021 dans un élevage de poules pondeuses dans le département du Nord (BHVSI du 30/11/2021). Au total, sur la saison 2021-2022, le pays a enregistré 1 378 foyers en élevages de volailles, 35 en basses-cours et 63 cas sauvages entre le 08/11/2021 et le 22/06/2022, selon le site du ministère (lien). Le sous-type H5N1 représente plus de 95% des foyers et cas : c’est le seul sous-type identifié à partir du 09/11/2022, à l’exclusion des virus pour lesquels le sous-type de neuraminidase n’a pu être déterminé. Plusieurs épizooties consécutives ont été observées dans les élevages de volailles (Figure 1) :
- A partir du 14/12/2021 (Gers), première introduction en élevage de volailles dans le sud-ouest (Gers, Hautes-Pyrénées, Landes et Pyrénées-Atlantiques) rapidement suivie par une diffusion du virus entre les élevages de la zone jusqu’à mi-février 2022 avec un total de 365 foyers de volailles sur l’ensemble de la saison (source : ADIS le 27/06/2022)
- A partir du 01/01/2022 (Vendée), première introduction en élevage de volailles dans la zone Ouest (Régions Bretagne, Normandie et Pays de la Loire et département des Deux-Sèvres) suivie de la détection de foyers sporadiques puis d’une augmentation brutale du nombre de foyers par diffusion intense entre les élevages de la zone de mi-février à fin avril 2022, avec un total de 861 foyers domestiques sur l’ensemble de la saison (source : ADIS le 27/06/2022)
- A partir du 10/03/2022 (Cantal), introduction en élevage de volailles dans la zone Centre-Ouest (Aveyron, Cantal, Charente, Corrèze, Dordogne, Haute-Vienne, Lot, Lot-et-Garonne) suivie par une circulation du virus dans les élevages de cette même zone jusqu’à mi-mai 2022, pour un total de 137 foyers domestiques pendant l’ensemble de la saison (source : ADIS le 27/06/2022)
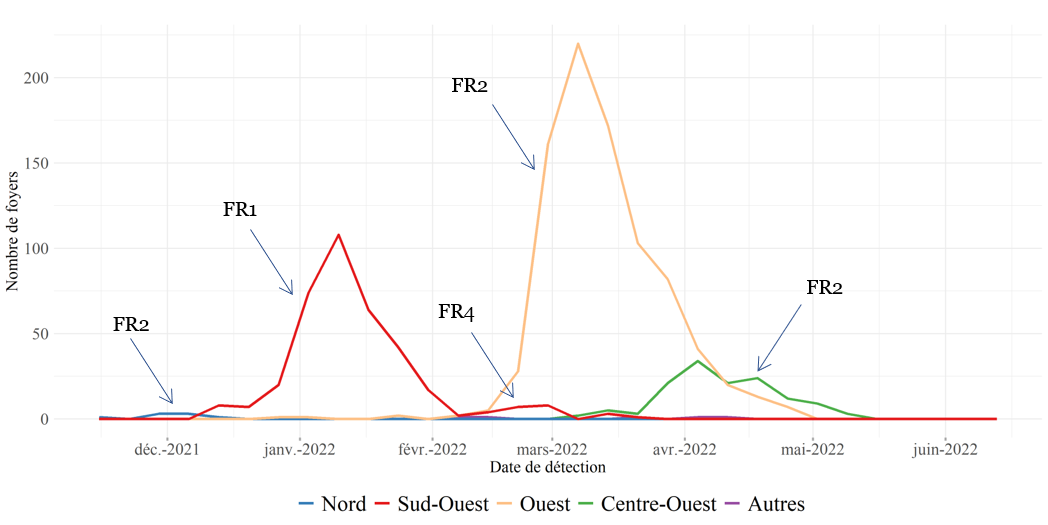
Figure 1. Nombre hebdomadaire de foyers de « volailles » détectés en France durant la saison 2021-2022 entre le premier foyer détecté le 21/11/2021 et le 19/06/2022, par aire de répartition géographique. Les génotypes majoritaires associés aux pics épizootiques de chacune des quatre régions spécifiquement identifiées sont mentionnés : les virus de génotype FR2 identifiés dans la zone Ouest puis dans la zone Centre-Ouest sont directement apparentés entre eux. Toutefois, le jeu de séquences obtenues n’étant ni exhaustif ni représentatif, par construction, de l’intégralité des foyers, un biais de sélection peut avoir orienter l’identification des associations représentées ici, entre un génotype et un pic épizootique donné (source : Commission européenne ADIS le 20/06/2022, LNR). NB : les données des dernières semaines sont à interpréter avec précaution, compte tenu des délais entre confirmation et notification.
|
Nom du cluster de foyers domestiques |
Régions ou départements concernés |
|
Nord |
Région Hauts-de-France |
|
Sud-Ouest |
Départements du Gers, des Hautes-Pyrénées, Landes et Pyrénées-Atlantiques |
|
Ouest |
Régions Bretagne, Normandie et Pays de la Loire et département des Deux-Sèvres |
|
Centre-Ouest |
Départements de l’Aveyron, Cantal, Charente, Corrèze, Dordogne, Haute-Vienne, Lot, Lot-et-Garonne |
|
Autres |
Autres (Indre-et-Loire et Loir-et-Cher) |
Les analyses phylogénétiques de 415 séquences de génomes complets de virus IAHP A(H5N1), issues de cas en avifaune sauvage ou chez des oiseaux captifs et de foyers en élevages de volailles détectés depuis novembre 2021 ont été réalisées. Elles montrent une grande diversité qui révèle que de nombreuses introductions ont eu lieu sur l’ensemble du territoire. Au moins huit génotypes de virus A(H5N1) ont en effet été détectés. Ils ont tous été détectés dans l’avifaune sauvage et pour quatre d’entre eux également dans des élevages de volailles.
Le premier génotype, nommé arbitrairement FR1, a été détecté sur une grande partie du territoire français et tout au long de l’hiver 2021-2022 chez des oiseaux sauvages, captifs ou domestiques. Il a notamment été détecté chez des oiseaux sauvages ou captifs de la Meuse, de l’Ain, de la Marne, de l’Aude, des Côtes-d’Armor, de la Nièvre, de la Loire, de Seine-Maritime et de la Loire-Atlantique. Ce génotype a été identifié majoritairement dans les élevages du sud-ouest de la France (Gers, Pyrénées-Atlantiques, Landes et Hautes-Pyrénées). Il a également été détecté sporadiquement dans des élevages de Vendée (début janvier 2022), de l’Indre et de l’Indre-et-Loire. Ce génotype FR1 avait par ailleurs été retrouvé en Italie, République tchèque, Pologne, Russie, Estonie et Angleterre depuis septembre 2021.
Le second génotype FR2 correspond à des séquences provenant d’élevages de volailles des départements du nord et de l’ouest de la France, d’une basse-cour du Morbihan ainsi que d’oiseaux sauvages de l’Ain, de l’Aube et de la Seine-Maritime. L’étroite parenté des premiers génomes viraux appartenant au génotype FR2 détectés à partir de fin janvier 2022 dans de nombreux élevages de Vendée indique un lien direct ou très proche entre ces virus. À partir de la mi-mars 2022, ce génotype a été détecté dans des élevages en Bretagne et dans la zone Centre-Ouest. Ce génotype avait également été identifié depuis septembre 2021 en Russie, aux Pays-Bas, en Belgique et en Croatie.
Le troisième génotype FR3 a été uniquement détecté chez des oiseaux sauvages de l’Ain, où les génotypes FR1 et FR2 ont également été détectés, sur une période courte (de mi-décembre 2021 à début janvier 2022). Ce génotype n’a par ailleurs été rapporté qu’en Italie.
Le quatrième génotype FR4 a été détecté à une dizaine de reprises en France, les premières fois chez une oie cendrée sauvage dans les Hautes-Pyrénées fin décembre 2021 et dans une basse-cour de Haute-Garonne, environ 10 jours plus tard. Il a également été détecté plus récemment, à partir de début février 2022, dans des élevages des Hautes-Pyrénées et du Gers. Ceci suggère une circulation à bas bruit de ce génotype dans cette région pendant presque deux mois, probablement dans l’avifaune sauvage. Aucune séquence européenne apparentée n’est actuellement décrite dans les banques de données.
Les génotypes FR5, FR6, FR7 et FR8 n’ont pour l’instant été détectés que sporadiquement en France respectivement chez des oiseaux sauvages en Moselle, dans les Côtes-d’Armor, dans la Marne et dans un élevage de Mayenne.
Dans les élevages du sud-ouest de la France (Gers, Pyrénées-Atlantiques, Landes et Hautes-Pyrénées), au moins deux génotypes distincts ont circulé : FR1 et FR4. Le génotype FR1 a très majoritairement diffusé entre élevages de mi-décembre 2021 à janvier 2022 et la reprise des détections à partir de mi-février 2022 dans les Hautes-Pyrénées et le Gers est associée à l’autre génotype, FR4. Une analyse phylogénétique plus fine réalisée à partir des données actuellement disponibles suggère également qu’au moins cinq introductions du génotype FR1 dans les élevages de volailles du Sud-Ouest ont eu lieu à partir de la mi-décembre 2021 :
Le premier groupe phylogénétique rassemble des séquences obtenues sur des échantillons provenant d’élevages du Gers.
Le second groupe phylogénétique rassemble des séquences obtenues sur des échantillons provenant d’élevages des Landes et des Pyrénées-Atlantiques (communes d’Hastingues et Came).
Les séquences virales identifiées dans les communes de Cazalis, Malaussanne et d’autres communes voisines forment un groupe phylogénétique suffisamment différent du groupe précédent pour suggérer une troisième introduction de virus IAHP A(H5N1) dans le sud-ouest de la France. Ce troisième groupe phylogénétique a été le plus fréquemment détecté dans le sud-ouest de la France : il est principalement présent dans le sud-est des Landes où il a d’abord été détecté puis a diffusé vers l’est, le nord-ouest et le sud-est et a également atteint le Gers, les Pyrénées-Atlantiques et les Hautes-Pyrénées.
Un quatrième groupe phylogénétique du génotype FR1 a été détecté dans le département des Pyrénées-Atlantiques (communes de Dognen, Poey-d’Oloron et Aren) ainsi que dans le département des Landes (commune de Borderes-et-Lamensans). L’étroite parenté des génomes viraux de ce groupe phylogénétique suggère qu’un lien direct entre les élevages touchés par ce virus est plus que probable.
Un dernier groupe phylogénétique du génotype FR1 a été identifié dans les communes d’Orthez et Sallespisse dans les Pyrénées-Atlantiques, ainsi que dans le Tarn. Les distances génétiques observées ici suggèrent également un lien probable entre ces élevages.
Dans les élevages du Grand-Ouest de la France (régions Pays de la Loire, Bretagne et département des Deux-Sèvres), au moins deux génotypes distincts ont circulé : FR1 et FR2. Une analyse phylogénétique plus fine réalisée à partir des données actuellement disponibles suggère qu’au moins trois introductions du génotype FR1 et une introduction du génotype FR2 ont eu lieu.
Le génotype FR1 a été détecté uniquement de manière sporadique de début janvier à février 2022 en Loire-Atlantique (oiseaux sauvages) et en Vendée et Deux-Sèvres (élevages de volailles) : du fait des distances génétiques observées, il n’y a pas de lien direct avec les virus de génotype FR1 détectés dans le Sud-Ouest.
Le génotype FR2 a été à l’origine de l’explosion des cas observés dans le Grand-Ouest à partir de fin février 2022, à partir d’une ou plusieurs introductions initiales dans le nord-ouest de la Vendée, suivies d’une transmission inter-élevages liée à l’activité humaine vers les départements limitrophes (Loire-Atlantique, Maine-et-Loire et Deux-Sèvres). Les analyses phylogénétiques indiquent également que les séquences identifiées dans quatre élevages de Bretagne en mars 2022 sont proches des séquences identifiées dans la région des Pays de la Loire : cela conforte, pour l’un des foyers en Bretagne, l’hypothèse principale de contamination identifiée par l’enquête épidémiologique qui est l’introduction du virus via le transfert de canetons contaminés. Pour les autres foyers détectés en Bretagne, une introduction par l’avifaune sauvage infectée (relais avec les zones contaminées des Pays de la Loire) peut être suspectée, en l’absence d’autres liens épidémiologiques constatés.
L’analyse phylogénétique de 37 séquences de génomes viraux complets détectés dans la zone Centre-Ouest indique qu’elles sont toutes regroupées en un seul cluster phylogénétique du génotype FR2 et sont directement apparentées aux séquences identifiées dans la zone du Grand-Ouest, suggérant donc une seule source d’introduction dans la zone Centre-Ouest à partir de la zone Grand-Ouest. À ce stade, plusieurs hypothèses d’introduction du virus dans cette zone restent envisageables : via l’avifaune sauvage et via les activités humaines. Le scénario le plus probable de la diffusion du virus IAHP H5N1 de génotype FR2 dans cette zone est donc une introduction primaire dans la zone index (frontière Cantal/Lot) suivie d’une diffusion inter-élevages (par voie aérienne, mouvements ou contacts indirects liés aux activités d’élevage).
En conclusion, la saison 2021-2022 est marquée en France par un nombre élevé de génotypes appartenant à un seul sous-type de virus IAHP A(H5N1), dont deux génotypes associés à chacune des vagues épizootiques majeures en élevages de volailles (Figure 1) : FR1 majoritaire dans le Sud-Ouest (de mi-décembre 2021 à début février 2022), puis FR2 majoritaire dans l’Ouest (de mi-février à mi-mai 2022). A titre de comparaison, seuls trois génotypes correspondants chacun à un sous-type différent de virus IAHP avaient été détectés durant la saison 2020-2021 (source : LNR le 10/06/2022).
[1] Laboratoire de référence de l’Union Européenne